
PMAT是一款基于三代测序数据(HiFi/CLR/ONT)的植物线粒体图形化基因组组装工具包。除了植物线粒体基因组,PMAT还适用于叶绿体图形化基因组的组装。目前,PMAT仅支持在Linux系统上运行,源码已公布在Github。大家在使用过程中遇到问题,可以添加文章底部的开发者微信或关注微信公众号:有趣儿的生信。
使用说明
安装
方法一:使用git安装
1
2
3
4
git clone https://github.com/bichangwei/PMAT.git
cd PMAT/bin
chmod a+x PMAT
PMAT --help
方法二:源代码安装
1
2
3
4
5
wget https://github.com/bichangwei/PMAT/archive/refs/tags/v1.5.2.tar.gz
tar -zxvf v1.5.2.tar.gz
cd PMAT-1.5.2/bin
chmod a+x PMAT
PMAT --help
依赖环境
- BLASTn:需要安装并添加到环境变量中;
- Singularity:当PMAT版本低于v1.3.0需要安装singularity,Singularity安装方法可以点击;
- Apptainer:当PMAT版本大于等于v1.3.0需要安装Apptaineris,具体安装方法可以点击;
- Canu 和 NextDenovo:对于测序数据为CLR和ONT需要安装Canu(>= v2.0)或者NextDenovo
参数介绍
执行PMAT --help 查看PMAT的参数信息,主要包括两个功能:一步法组装(autoMito)和手动构建组装图(graphBuild)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
usage: PMAT <command> <arguments>
______ ___ __ ____ _____________
| __ \ | \ / | / __ \ |_____ _____|
| |__) | | |\ \ / /| | / / \ \ | |
| ____/ | | \ \ / / | | / /____\ \ | |
| | | | \ \ / / | | / /______\ \ | |
| | | | \ \/ / | | / / \ \ | |
|__| |_| \__/ |_| /_/ \_\ |_|
PMAT An efficient assembly toolkit for plant mitochondrial genome
Version 1.5.2
Contributors Bi,C. and Han,F.
Email bichwei@njfu.edu.cn, hanfc@caf.ac.cn
For more information about PMAT, please see https://github.com/bichangwei/PMAT
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version and exit
Commands:
autoMito One-step de novo assembly of the mitochondrial genome.
This command can generate the master assembly graph
from raw sequencing data directly.
graphBuild If 'autoMito' mode fails to generate the mitogenome
assembly graph, you can use this command to manually
select seeds for assembly.
** autoMito 一步法组装线粒体图形化基因组
autoMito
autoMito功能主要有以下参数:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
Required arguments:
-i INPUT, --input INPUT
input raw sequencing file
-o OUTPUT, --output OUTPUT
output directory
-st SEQTYPE, --seqtype SEQTYPE
sequencing platform(ONT/CLR/HiFi)
-g GENOMESIZE, --genomesize GENOMESIZE
Please enter the genome size of the species, such as 1G, 1000M.
optional arguments:
-h, --help show this help message and exit
-tk TASK, --task TASK
all/p1/ Default: all
all : De novo assembly including error correction for ONT/CLR data and no error correction for HiFi data
p1 : Import error-corrected ONT/CLR data for direct assembly
-tp TYPE, --type TYPE
mt/pt/all Default: mt
mt : Assembling the mitochondrial genome
pt : Assembling the chloroplast genome
all : Assembling the mitochondrial and chloroplast genomes
-cs CORRECTSOFT, --correctsoft CORRECTSOFT
Correcting software using nextDenovo or Canu. Default: NextDenovo
-cp CANU, --canu CANU
Please provide the install path of canu.
-np NEXTDENOVO, --nextDenovo NEXTDENOVO
Please provide the install path of nextDenovo.
-cfg CORRECTCFG, --correctcfg CORRECTCFG
config file for nextdenovo correct
-fc FACTOR, --factor FACTOR
Subset extraction of error-corrected ONT, CLR or HiFi data. Sampling ratio factor in 0-1. Default: 1
-sd SUBSEED, --subseed SUBSEED
Sampling set random number seeds, Default: 6
-bn BREAKNUM, --breaknum BREAKNUM
break long reads (>30k) with this. Default: 20000
-ml MINOVERLAPLEN, --minoverlaplen MINOVERLAPLEN
set minimum overlap length. Default: 40
-mi MINIDENTITY, --minidentity MINIDENTITY
set minimum overlap identification. Default: 90
-cpu CPU The number of threads. Default: 8
-l MINLINK, --minLink MINLINK
Filter according to the minimum link depth provided by the user
-m, --mem Flag to keep sequence data in memory to speed up cup time
-u, --unloop Flag for attempting automatic unloop
-v, --version show program's version and exit
autoMito功能的使用说明:
| 参数 | 说明 (autoMito需要安装Blastn,并添加到环境变量中) |
|---|---|
-i |
输入三代测序数据。数据格式支持fastq、fasta和以.gz结尾的压缩文件 |
-o |
输出文件路径 |
-st |
声明测序数据的格式,可以是HiFi、ONT或者CLR |
-g |
声明基因组大小,如1g,1000m,1000kb |
-tk |
该参数有两个选项,分别是all(默认)和p1。 当输入测序数据为ONT和CLR时,autoMito将对测序数据进行纠错(Canu或者nextDenovo)并修剪(Canu)。如果输入数据为完成纠错的高质量reads可以使用p1跳过纠错和修剪的过程。如果测试数据类型为HiFi可以忽略该参数 |
-tp |
该参数有三个选项,分别是mt(默认)、pt和all,用于指定组装细胞器类型 |
-cs |
该参数针对ONT和CLR数据类型而设计,用于指定纠错软件。该参数可选 nextDenovo(默认)和 Canu(>= v2.0) |
-cp |
该参数用于指定canu的安装路径,如/home/user/path/canu,如果使用未纠错的ONT和CLR数据时该参数为必选参数 |
-np |
该参数用于指定nextDenovo的安装路径,如/home/user/path/nextDenovo,如果使用未纠错的ONT和CLR数据,并且-cs指定nextDenovo纠错,该参数为必选参数 |
-cfg |
该参数用于指定nextDenovo的配置文件,如果-cs指定nextDenovo,该参数为必选参数。配置文件的设置推荐查看NextDenovo |
-fc |
该参数用于指定用于组装过程的数据量,如果-fc指定为0.5,则随机选择一半测序数据进行组装,默认为1 |
-ml |
该参数用于指定映射使用的重叠最小长度,推荐设置: 40~200(默认:40) |
-mi |
该参数用于指定映射使用的最小重叠百分比,推荐设置: 90~98(默认:90) |
-cpu |
该参数用于指定使用的线程数 |
-u |
是否进行自动解环 |
** graphBuild 手动构建组装图
graphBuild
当autoMito功能无法正确输出结果时,可以使用graphBuild手动指定种子进行图形化线粒体基因组组装
graphBuild 功能主要有以下参数:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
Required arguments:
-c CONTIGGRAPH, --ContigGraph CONTIGGRAPH
PMATContigGraph.txt: a file that can get all connections between contigs.
-a ALLCONTIGS, --AllContigs ALLCONTIGS
PMATAllContigs.fna: a file that can get all the information of contigs.
-o OUTPUT, --output OUTPUT
output directory
-gs GENOMESIZE, --genomesize GENOMESIZE
Please enter the genome size of the species, such as 1G, 1000M.
-rs READSIZE, --readsize READSIZE
The read size or file for assembly, such as 5G or assembly_seq.cut20K.fasta.
optional arguments:
-h, --help show this help message and exit
-tp TYPE, --type TYPE
mt/pt/all Default: mt
mt : Assembling the mitochondrial genome
pt : Assembling the chloroplast genome
all : Assembling the mitochondrial and chloroplast genomes
-cpu CPU The number of threads. Default: 8
-s SEEDS [SEEDS ...], --seeds SEEDS [SEEDS ...]
ContigID for extending. Multiple contigIDs should be separated by space. For example: 1 312 356
-u, --unloop Flag for attempting automatic unloop
-l MINLINK, --minLink MINLINK
Filter according to the minimum link depth provided by the user
-v, --version show the program's version number and exit
graphBuild功能的使用说明:
| 参数 | 说明 (graphBuild需要安装Blastn,并添加到环境变量中) |
|---|---|
-c |
该参数用于指定autoMito命令输出的结果文件PMATContigGraph.txt |
-a |
该参数用于指定autoMito命令输出的结果文件PMATAllContigs.fna |
-gs |
声明基因组大小,如1g,1000m,1000kb |
-rs |
声明用于组装数据量,如10g,10000m等,或者指定autoMito命令输出的结果文件assembly_seq.cut20K.fasta |
-tp |
该参数有三个选项,分别是mt(默认)、pt和all,用于指定组装细胞器类型 |
-cpu |
该参数用于指定使用的线程数 |
-s |
手动选择用于指定延伸的seeds,推荐指定三个以上的seeds,并使用空格分隔,如:1 312 356。seeds的选择可以结合PMATContigGraph.txt中每个contig的深度判断 |
示例
- 下载拟南芥的HiFi测试数据集:
1
wget https://github.com/bichangwei/PMAT/releases/download/v1.1.0/Arabidopsis_thaliana_550Mb.fa.gz
- 运行autoMito进行一步法组装线粒体基因组:
1
PMAT autoMito -i Arabidopsis_thaliana_550Mb.fa.gz -o ./test1 -st hifi -g 120m -m
- 当autoMito无法自动获得组装图GFA文件时,可以根据PMATContigGraph.txt中每个contigs深度选择潜在的线粒体基因组contig作为seeds,推荐seeds数量大于3个:
1
PMAT graphBuild -c ./test1/assembly_result/PMATContigGraph.txt -a ./test1/assembly_result/PMATAllContigs.fna -gs 125m -rs ./test1/subsample/assembly_seq.cut20K.fasta -o ./test1_gfa -s 343 345 905 513 1344
- PMAT不同线程下组装时间结果:
1
8 CPUs: 13m25.342s; 16 CPUs: 9m29.853s; 32 CPUs: 8m42.429s; 64 CPUs: 7m57.279s
- 下载苹果的HiFi测试数据集:
1
wget https://github.com/bichangwei/PMAT/releases/download/v1.1.0/Malus_domestica.540Mb.fasta.gz
- autoMito进行一步法组装:
1
PMAT autoMito -i Malus_domestica.540Mb.fasta.gz -o ./test3 -st hifi -g 703m -m
- graphBuild进行手动构建组装图:
1
PMAT graphBuild -c ./test3/assembly_result/PMATContigGraph.txt -a ./test3/assembly_result/PMATAllContigs.fna -gs 225m -rs ./test3/subsample/assembly_seq.cut20K.fasta -o ./test3_gfa -s 1 2 15391
- 不同线程下PMAT运行时间比较:
1
8 CPUs: 21m12.306s; 16 CPUs: 12m14.663s; 32 CPUs: 7m58.749s; 64 CPUs: 6m48.915s
- 下载菜豆CLR测序数据:
1
ascp -v -QT -l 400m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR291/006/SRR2912756/SRR2912756_subreads.fastq.gz .
- 运行autoMito进行一步法组装线粒体基因组,这里使用nextDenovo进行纠错,canu对纠错后的数据进行修剪:
1
PMAT autoMito -i SRR2912756_subreads.fastq.gz -o ./test_clr -st clr -g 540m -cs nextDenovo -np path/nextDenovo -cp path/canu -cfg nextdenovo.cfg -m
- 下载美洲黑杨ONT测序数据:
1
ascp -v -QT -l 400m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR122/038/SRR12202038/SRR12202038_1.fastq.gz .
- 运行autoMito进行一步法组装线粒体基因组,这里使用canu进行纠错并修剪:
1
PMAT autoMito -i SRR12202038_1.fastq.gz -o ./test_ont -st ont -g 430m -cs canu -cp path/canu -m
输出结果
- 输出文件包括:
*/subsample/assembly_seq_subset.1.0.fasta, 用于组装的数据集;*/subsample/assembly_seq.cut20K.fasta, 对组装的数据集进行截断;*/assembly_result/PMATAllContigs.fna, 包含contigs序列的组装结果文件;*/assembly_result/PMATContigGraph.txt, 包含contigs连接关系的组装结果文件;*/assembly_result/PMAT_mt_raw.gfa, 线粒体基因组初始组装图;*/assembly_result/PMAT_mt_master.gfa, 优化后的线粒体基因组组装图;*/assembly_result/PMAT_pt_raw.gfa, 叶绿体基因组初始组装图;*/assembly_result/PMAT_pt_master.gfa, 优化后的叶绿体基因组组装图
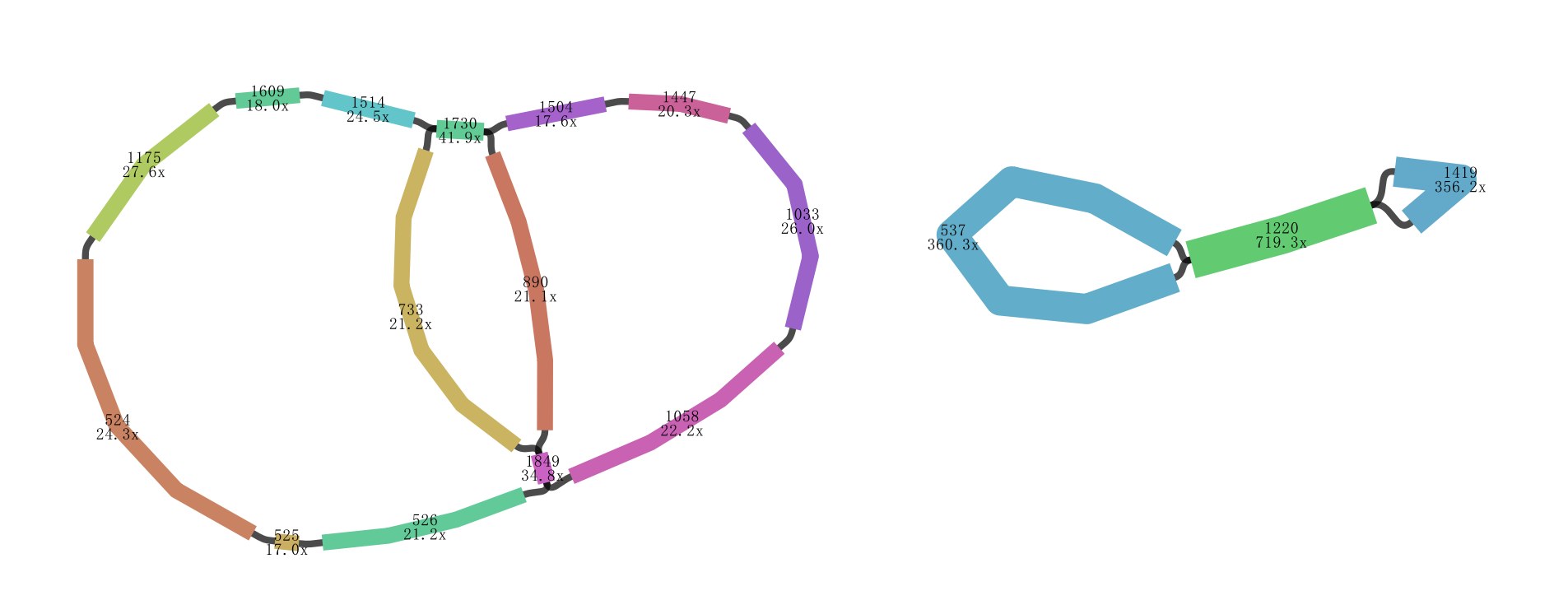
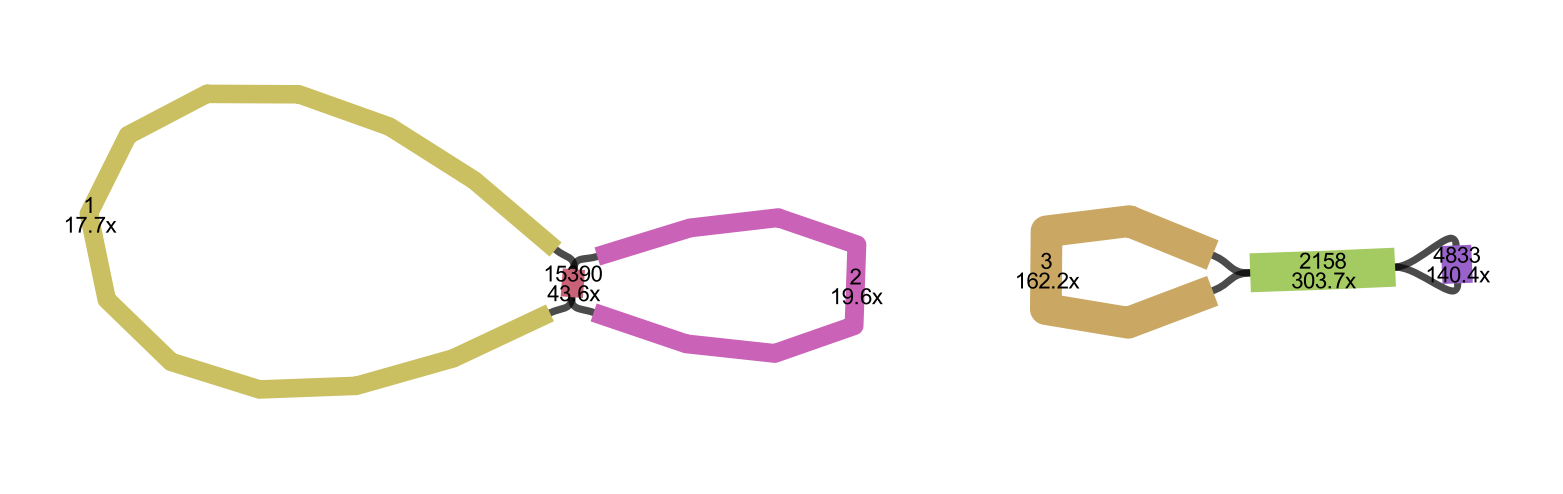
** 注意优化后的master.gfa并不一定比raw.gfa有更好的结果,可以使用Bandage对结果进行查看:
Arabidopsis thaliana
Malus domestica
引用PMAT
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., Xu, L., He, W., Wu, Z., Yin, T. (2024). PMAT: an efficient plant mitogenome assembly toolkit using ultra-low coverage HiFi sequencing data. Horticulture Research, Accepted.
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 13:914635.doi:10.3389/fpls.2022.914635.
** 无法进入群聊可以添加下方作者的联系方式,请备注姓名+单位+PMAT
联系作者
毕长伟, 南京林业大学,邮箱: bichwei@njfu.edu.cn; 微信号: (13770664619 )
韩富川, 中国林业科学院亚热带林业研究所,邮箱: hanfc@caf.ac.cn; 微信号: (h18437901321)
© 版权声明: 本博客所有文章除特别声明外,著作权归作者所有。转载请注明出处!